The "BLAST" tool allows users to align their query sequences or FASTA files with the MIrROR database. Since the server has limited resources, performing BLAST with more than 10 sequences may take a long time.
Accessing the BLAST Tool
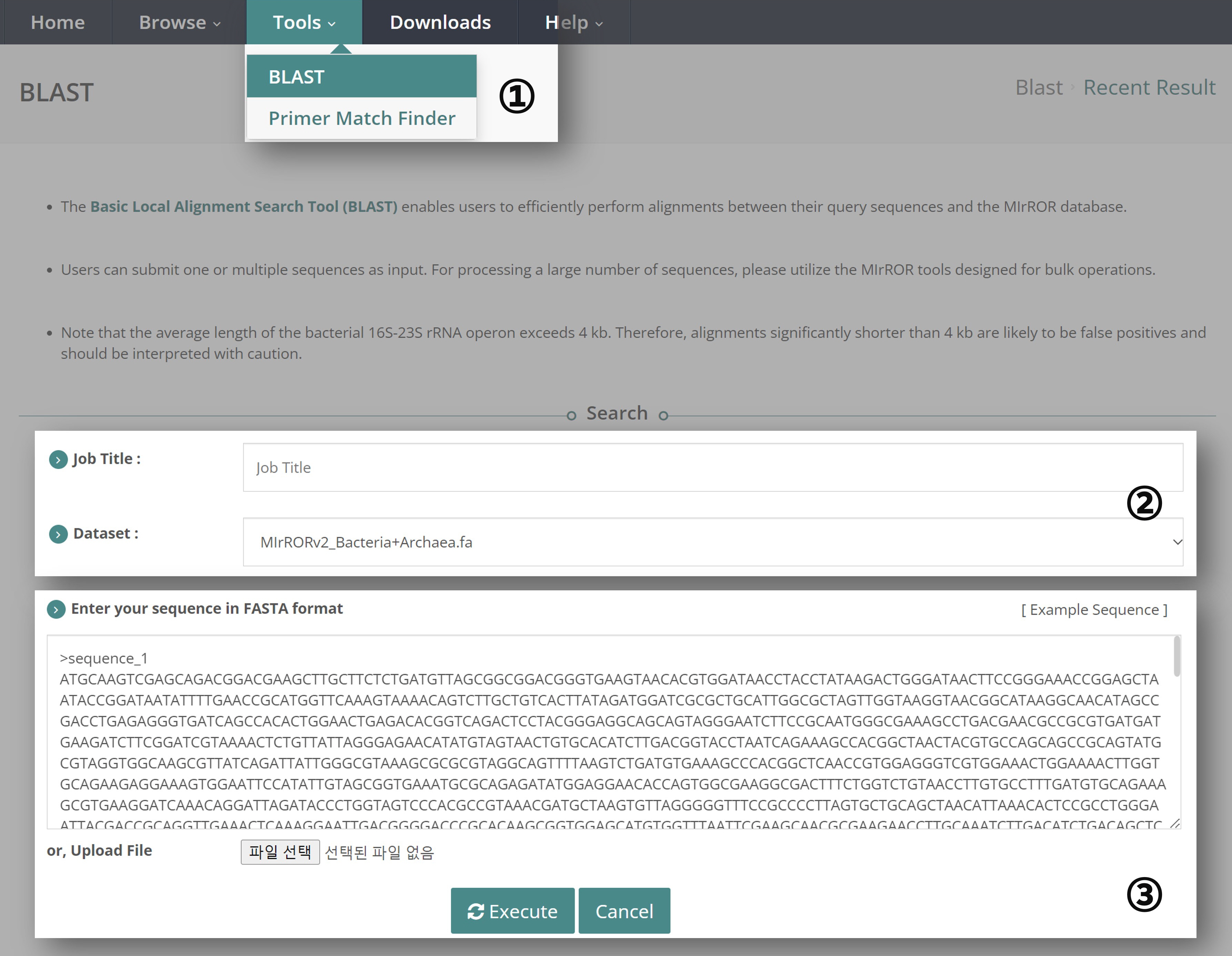
① Navigate to the "Tools" tab and select "BLAST".
② Enter the "Job Title". The Dataset for BLAST is pre-set to the integrated MIrROR Database, which includes both Bacteria and Archaea.
③ Users can perform BLAST by entering query sequences directly or by uploading a FASTA file. Only the FASTA format is supported. If you are not familiar with the FASTA format, please click ‘Example sequence’. Once the sequence is ready, click the ‘Execute’ button.
Viewing BLAST Results

④ The information about the genome is summarized in pie charts:
- Job Title: The title of the BLAST job.
- RID: Request ID for retrieving results from a specific NCBI BLAST search.
- Results for: If there are two or more sequences, you can select one to view.
- Query Length: The length of the query sequence.
- Program: The algorithm and version of the BLAST search.
- Database Count / Length: The number of sequences in the database and the total number of base pairs.
- Kappa | Lambda | Entropy: Statistical details about the BLAST search, including Kappa, lambda, and entropy values.
⑤ The BLAST results are shown in a format similar to the ‘BLASTn output format 6’:
- Operon Sequence: Subject sequence ID.
- Bit Score: The bit score of the alignment.
- Alignment Length: The length of the alignment.
- Mismatches: The number of mismatches in the alignment.
- Query Cover: The coverage of the query sequence per subject.
- Percent Identity: The percentage of identical matches.
- E Value: The expect value of the alignment.
- GTDB Taxonomy: The taxonomic name of the subject sequence based on GTDB.
Alignment Details

⑥ Users can check the alignment between the query sequence and the MIrROR database in bit score order.
